»Volume 2014 Issue 02 (May)
Analysis of Phosphine in Dried Foodstuffs via Headspace-GC-MSD
Roland Perz, Anne Benkenstein, Helmut Köbler, Ellen Scherbaum, Dieter Köhl, Anja Barth and Michelangelo Anastassiades
Chemisches und Veterinäruntersuchungsamt Stuttgart
Schaflandstraße 3/2
70736 Fellbach, Germany
Phone: +49 711 3426 1941
Fax: +49 711 3426 1149
Email: Roland.Perz@cvuas.bwl.de
Keywords:
Phosphine, Headspace GC MS, fumigant, fumigation, environmental
Download as » PDF (1,141 KB)
Abstract
Phosphine is one of the most widely used, cost-effective and rapidly acting fumigants. In EU legislation , maximum residue limits for the sum of phosphine and phosphides in foodstuff are set to within a range of 0.01 and 0.1 mg kg-1, depending on the commodity. A highly sensitive headspace-GC-MSD method was developed achieving limits of quantitation as low as 0.1 µg kg-1; this enabled not only the monitoring of MRLs, but also the exposure of improper applications. In all, 115 samples of dried foodstuff from the local market such as cereals, nuts, and legumes were analyzed for phosphine residues. Of these, 35 samples contained phosphine in amounts exceeding 0.1 µg kg-1, while 14 samples (12 % of all) exceeded 1 µg kg-1. Interestingly, seven of these 14 samples were labeled as being from organic production, where phosphine application is not allowed. Monitoring activities will be continued.
Introduction
Globalization has led to an increased trade of goods between countries from different continents. In 2011 maritime trade was estimated at 500 million containers, transporting goods from all parts of the world [1]. However, stowaways such as pests are inevitably carried along with the goods as well. Fumigation of containers is common practice in the export and import of foods, both in order to preserve the foods during the long trip and to eliminate any pests that could be brought into a country with the food. Methyl bromide was previously among the most widely used fumigants but its production and use was restricted by the Montreal Protocol due to its role in ozone depletion [2]. Nowadays phosphine (PH3) is one of the most widely used, cost-effective and rapidly acting fumigants not expected to leave higher residues on treated products. Cases of pests developing resistance to phosphine, however, have been reported from different parts of the world.
Properties of Phosphine and Phosphides
Phosphine is a colorless and odorless, flammable gas. Typical impurities (e.g. diphosphane P2H4) cause an odor of garlic or decaying fish. Furthermore, traces of diphosphane increase the risk of self-ignition. Phosphine affects the central nervous system and irritates the lungs. It is also considered very toxic for fish. Overexposure of humans to phosphine leads to symptoms such as nausea, vomiting, numbness and spasms. Lethal intoxications have also been reported. Chronic poisonings are not noted, however, because minor doses are constantly detoxicated in the blood [3].
In dry conditions aluminum, magnesium and zinc phosphide are stable crystals, but when they come in contact with moisture from crops, soil or air they gradually release phosphine.
Legal Aspects
In Germany several products containing phosphine and its salts aluminum phosphide and magnesium phosphide are registered for use on coffee, cocoa, oily seeds, dried fruit, legumes and stored cereals (Federal Office of Consumer Protection and Food Safety, BVL). Zinc phosphide is permitted, furthermore, as a rodenticide in the form of pellets.
In EU legislation , maximum residue limits for phosphine and phosphides in foodstuff range from 0.01 to 0.1 mg kg-1, depending on the commodity (Reg. (EC) No 149/2008) [4].
Annex 2 of Commission Regulation (EC) 889/2008 [5] contains a restricted list of products and substances which may be used in organic farming for various purposes including plant protection, cleaning and disinfection (see Article 16 of Council Regulation (EC) No 834/2007 [6]). Phosphine and phosphides are not listed. Article 26 of Commission Regulation (EC) 889/2008 stipulates that every measure be taken to avoid cross contamination of organically grown products with conventional products, prescribing the separate storage and suitable cleaning of production equipment. Therefore, phosphine residues should not principally be contained in organic food.
Analytical Approaches
Due to its high volatility phosphine is not amenable to common multi-residue methods for pesticide residue analysis in food; thus, special single residue methods have to be applied.
The earliest attempts at determining phosphine used derivatization with titrimetric or photometric methods [7, 8]. As instrumental analysis techniques became more sensitive and reliable, however, phosphine, and fumigants in general, were preferably analyzed by GC, following the injection of liquid extracts into packed columns initially connected to thermal conductivity detectors [9]. Later on, the more sensitive and selective flame photometric [10–12], thermionic [12, 13] and mass spectrometric detectors [14] were used. With the introduction of new techniques enabling highly reproducible sampling in the gas phase, new methods were developed. These employ the purge and trap approach, which involves offline analyte enrichment [11, 15–17], the automated headspace sampling approach [18], or the headspace-SPME approach [19]. An overview of these methods is given in the review of Desmarchelier [20].
In 2003 Amstutz et al. published a gas chromatographic method for the analysis of phosphine in dry commodities involving the addition of aqueous sulfuric acid to the sample, a preconditioning at 80 °C in a closed vessel, and a headspace sampling of the gas-phase above the sample. Detection was accomplished with a flame photometric detector [21]. The limits of quantitation (LOQs) achieved were very low, with reported concentrations in real samples varying between 0.3 µg kg-1 and 2.5 µg kg-1.
Based on this published method, we have set up a new method also involving headspace sampling and GC-analysis, but employing a mass spectrometric instead of a flame photometric detector to obtain additional diagnostic information.
Experimental
Chemicals and Standards
Sulfuric acid (concentrated) analysis grade was purchased from Merck (Darmstadt, Germany). The analytical standard phosphine (purity ≥ 99.9 %, 100 ppm and 10 ppm dilution in nitrogen) was obtained from Linde AG.
Apparatus
Samples were ground at room temperature using a Grindomix GM 200 knife mill by Retsch (Haan, Germany). For safe handling of calibration gases, Tedlar gas sampling bags Nr. 24633 from Supelco (Sigma-Aldrich, Germany) and gas tight syringes PN 1710 100 µL and PN 1001 1000 µL from Hamilton (Martinsried, Germany) were used. Analytical balances capable of weighing units down to 0.1 mg or 0.01 g were from Mettler-Toledo (Greifensee, Switzerland).
An Agilent GC-MSD system (Waldbronn, Germany), consisting of a 6890 GC and a 5973 MSD was used for analysis.
The system was equipped with a MPS2 sampler from Gerstel (Mülheim/Ruhr, Germany) with a headspace agitator unit and a 2.5 mL syringe. Further, a KAS 4 PTV with a cryo unit to be run with liquid nitrogen and capable of maintaining -80 °C served as the injection port. Suitable liners filled with Tenax were from Gerstel. Chromatographic separation took place on a Rt-Q-Bond PLOT column (30 m × 0.32 mm × 10 µm) from Restek (Bad Homburg, Germany). To prevent single, loose particles of the stationary phase from entering the ion source of the MSD, a restriction capillary (5 m × 0.25 mm) from Agilent (Waldbronn, Germany) was inserted between the PLOT column and the MSD using an appropriate column connector (Agilent, Waldbronn).
Sample Preparation
Coarse-granular commodities, such as nut kernels or legumes, were ground with a knife mill. Heat development was minimized by intervallic grinding. An amount of 1 g of the powdery homogenate was weighed into a headspace vial, 7 mL of water were added and the vial was closed with a rubber stopper and vigorously shaken. Subsequently, the vial was filled to a level of 15 mL with sulfuric acid 10 % and immediately sealed. In the case of granular commodities (e.g. whole grains), up to 3 g were weighed into the headspace vial and 5 % sulfuric acid solution was rapidly poured into the vials up to a level of 15 mL (using a small beaker). The vials were immediately closed and shaken.
Headspace-GC-MSD Analysis
The following injection settings were used for GC-MSD analysis: the agitator temperature was set at 80 °C, incubation time was 10 min, shaking speed was 500 rpm, and the shaking interval 5 s, followed by a 2 s break. The syringe temperature was set at 85 °C, the injection volume at 2000 µL, the draw speed at 200 µL s-1 and the injection speed at 500 µL s-1. PTV conditions were as follows:
-80 °C initial temperature with 1.0 min initial time, heating ramp to 150 °C with a rate of 12.0 °C min-1, hold time 2 min. Carrier gas flow (helium) was set at 2.2 mL min-1 in constant flow mode, split ratio was 5:1. The oven temperature program started at 35 °C with 3 min initial time, followed by two heating ramps (10 °C min-1 to 100 °C, then 35 °C min-1 to 200 °C) and a final time of 4 min, resulting in a total run time of 16.4 min. The transfer line temperature was set at 240 °C. The mass selective detector (ionization in EI mode, 70 eV) worked in SIM mode recording the ions m/z 31, 33 and 34 after a solvent delay of 4 min with a dwell time of 100 ms for each ion. To gain sufficient detector sensitivity, it was essential to use a tune file especially for very low masses.
Method validation
The chromatographic separation employed was shown to be selective enough to largely exclude any disturbances by oxygen, hydrogen sulfide or other small molecules in the same m/z range. The acceptable retention of phosphine (capacity factor near 3) and the stable retention times, combined with a good chromatographic resolution and the presence of two diagnostic ions, provided a high degree of certainty in the identification and quantification of phosphine in all tested sample types (see Fig. 2).
Calibration curves of procedural calibration standards were linear up to at least 50 µg kg-1, with good correlation coefficients (R2 > 0.99). Due to matrix-dependent signal quenching effects (see Fig. 1), external non-matrix-matched calibrations are not recommended for final quantification. They are suitable for screening purposes, however. For accurate quantification, positive samples have to be re-analyzed by the standard additions approach or at least calibrated against procedural calibration standards prepared on a similar matrix.
Relative standard deviation of replicate analyses (n = 10) of spiked matrix (unground millet, 1 g per vial) at one level (42 ng g-1) was 5.8 %, thus showing good repeatability of the procedure.
By moderately milling dry pulses with incurred residues we observed single cases of phosphine signals that were up to twice as high as those for unground samples (data not shown). In the case of cereals with aged residues we did not observe this effect. Moderate grinding of dried samples is thus recommended, as long as temperatures are kept low. This improves the homogeneity of the material (and therefore the reproducibility and accuracy of results) and leads in some cases to higher (and more correct) results.
Since spiking a blank matrix to achieve aged residues at a known level is impossible, the evaluation of result trueness (deviation from the real value) by means of common recovery experiments was not possible. We were, however, able to check our method in a ring test, organized by Amrein et al. [22], and obtained good results (absolute z-score < 2) for real samples in all but one case.
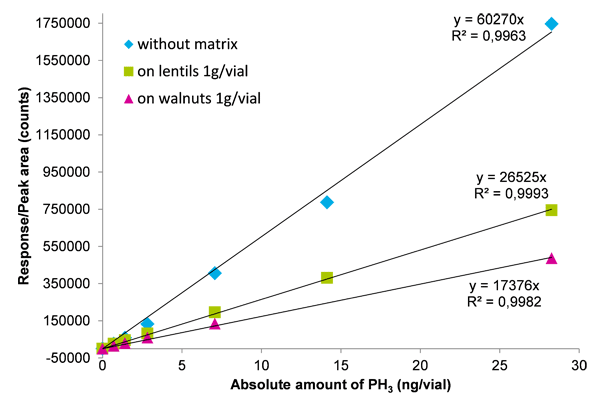
Results and discussion
Analytical Results and Method Adaptation
As discussed by Amrein et al. [22], there are several critical factors that have to be accounted for when implementing the method. Sample preparation is of core importance.
Grinding of the sample helps to improve homogeneity, but the decision to do so or not depends on the properties and condition of the commodities to be analyzed. In our milling experiments we have noticed single cases with remarkably higher signals following grinding. This might be due to better accessibility of the analyte, which probably penetrated the matrix over time. On the other hand, excessive grinding was intentionally used by Brockwell [10] to achieve complete PH3 release, so excessive grinding should be avoided. Interestingly, samples that were even older than 2 years having been stored in paper bags, which are definitely not gas tight, showed remarkable PH3 findings. The fact that PH3 signals are not sensitive to careful grinding and that residues of this very volatile analyte exist a long time leads to the assumption of tightly, but reversibly bound residues, which interact with matrix-components via either strong, non-covalent adsorption or covalent bonding. Covalent irreversible bonding has already been described by Berck for cereal samples [23] .
Some fluffy commodities such as dried herbs and bran tend to enclose air bubbles, thus causing a bias in the headspace volume within the vial. Instead of stirring to remove bubbles as proposed by Amrein [22], shaking with water and subsequent volume adjustment with higher concentrated acid was shown to be a good alternative. Reduced wettability, especially of powdery samples, can be overcome with this procedure, too.
Aside from sample homogeneity, reproducibility of results strongly depends on fast and experienced handling of liquid. The use of pipettes for filling the vials with acid was shown to be too slow, and lead to substantial losses of PH3.
We have also observed a significant loss of PH3 while sealed vials were sitting in the autosampler tray and concluded that, from the moment of sulfuric acid addition, no more than 4 hours should elapse before headspace sampling and injection. Thus, no more than 5 samples should be prepared at a time.
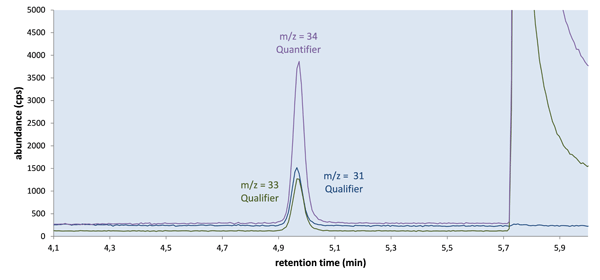
Amrein et al. [22] also discussed the influence of the sample amount on the results. Aside from signal optimization concerns, our goal was the safe handling of samples and method robustness. To reduce matrix-influence, we simply limited the maximum weight of powdery or much foam producing samples (like pulses) to 1 g, and of other samples to 3 g. We also refrained from using anti-foaming agents, as these might influence the liquid/headspace equilibrium of phosphine.
Results of Samples from the Local Market
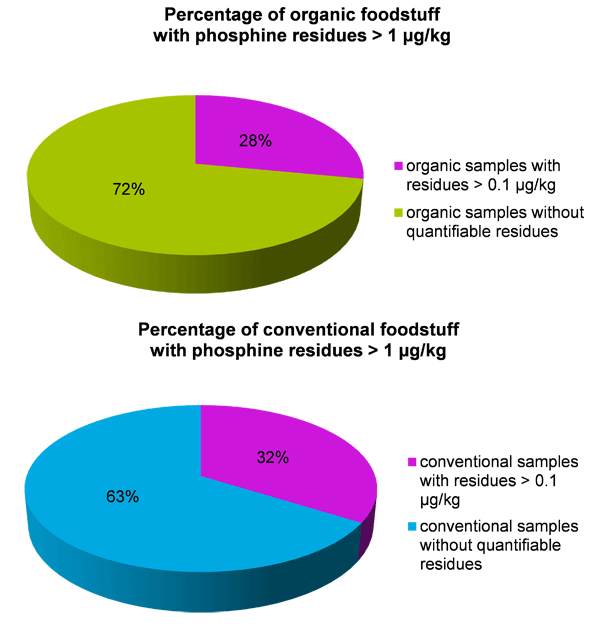
To get an overview of the residue situation, 115 samples of cereals, spices, oilseeds, legumes from conventional production and, to a smaller extent, from organic production were analyzed. In 38 of the tested samples residues of phosphine exceeded our LOQ of 0.1 µg kg-1. However, as expected, concentrations were well below the legal maximum residue limits. Fourteen out of 115 samples (12 % of all) contained residues exceeding 1 µg kg-1, see Table 1.

A detailed presentation of the results for conventional foodstuffs is found in Table 2 and, for organic foodstuffs, in Table 3.
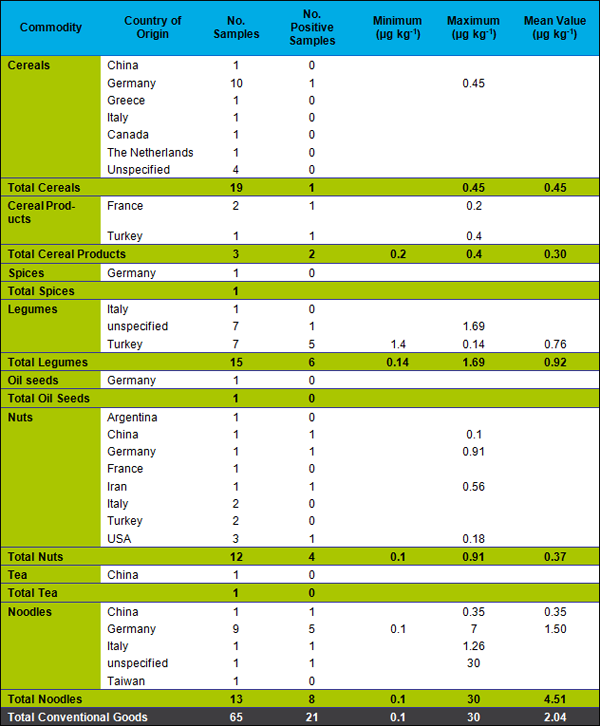
From the 21 conventional samples containing phosphine at levels exceeding our LOQ of 0.1 µg kg-1 7 were from Germany (thereof 5 noodle samples), 6 from Turkey, 2 from China, USA or not specified, respectively. One sample each originated in France, Italy, Iran and the USA. This indicates a widespread use of phosphine in many countries.
In contrast to fruits and vegetables, many other products do not require information regarding the country of origin on the packaging or price tag. Thus, in many cases, the country of origin for the samples analyzed was “unspecified”.
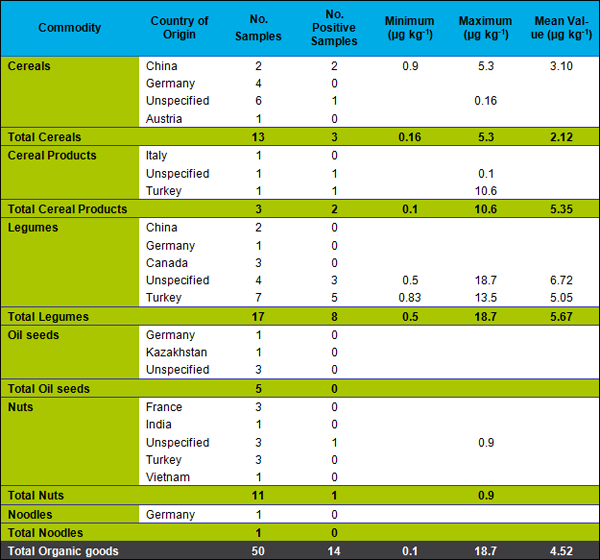
From the 14 organic samples containing quantifiable phosphine residues, 6 were from Turkey, 6 had an unknown origin and 2 originated in China.
Interestingly, processed foods such as noodles also frequently showed residues exceeding the LOQ. In these cases, the phosphine may have come from either the original ingredients (e.g. wheat, eggs) or from pest control measures taken during transport or storage of the finished products. In one case, we cooked a positive noodle sample to prepare the product as one would for a meal, and found afterwards that nearly half of the residual phosphine was still there. This indicates that, to some extent, phosphine residues can even survive exposure to boiling water.
Conclusion
Quantifiable concentrations of phosphine were found in 32 % of the conventional foodstuff samples, and in 28 % of the organic foodstuffs. As phosphine is not permitted for use in organic production and cross contamination should be minimized by appropriate measures, no residues should actually occur in organic products. The phosphine concentrations detected in conventional and organic products were in the same range. More research is needed to elucidate the reasons for the findings in organic products, potential options being cross-contamination, mingling of organic and conventional products and illegal applications. .
Residual phosphine can be bound tightly to the matrix and survive extended food storage or processing. Therefore, phosphine may occasionally occur in a broad variety of processed foods that have not yet been the focus of analytical chemists so far.
Acknowledgement
We would like to thank Mr. Thomas Amrein from Coop Swiss for his support and exchange of knowledge, as well as for the possibility to participate last-minute in a method validation ring test.
References
1. [Website] 2014; Available from http://www.hafen-hamburg.de/content/weltcontainerumschlag.
2. Montréal Protokol on Substances that Deplete the Ozone Layer. United Nations Environment Programme, 1995. Nairobi (Kenia).
3. [Website] 2013; Available from: http://www.umweltlexikon-online.de/RUBwerkstoffmaterialsubstanz/Phosphorwasserstoff.php.
4. COMMISSION REGULATION (EC) No 149/2008 of 29 January 2008 amending Regulation (EC) No 396/2005 of the European Parliament and of theCouncil by establishing Annexes II, III and IV setting maximum residue levels for products covered by Annex I thereto, in Official Journal of the European Union L58/1-3982008: Official Journal of the European Union. p. 398.
5. COMMISSION REGULATION (EC) No 889/2008 of 5 September 2008 laying down detailed rules for the implementation of Council Regulation (EC) No 834/2007 on organic production and labelling of organic products with regard to organic production, labelling and control, in Official Journal of the European Union L250/1, T.C.O.T.E. COMMUNITIES, Editor 2008.
6. Council Regulation (EC) No 834/2007 of 28 June 2007 on organic production and labelling of organic products and repealing Regulation (EEC) No 2092/91, in Official Journal of the European Union L189/1–23, T.C.O.T.E. UNION, Editor 2007: Official Journal of the European Union. p. 23.
7. Bruce, R.B., A.J. Robbins, and T.O. Tuft, Fumigant Residues, Phosphine Residues from Phostoxin-Treated Grain. Journal of Agricultural and Food Chemistry, 1962. 10(1): p. 18–21.
8. Kroeller, E., Determination of residual phosphine in foods. Deutsche Lebensmittel-Rundschau, 1968. 64(1): p. 6–9.
9. Berck, B., Determination of fumigant gases by gas chromatography. Journal of Agricultural and Food Chemistry, 1965. 113(4): p. 373–7.
10. Brockwell, C.A., Determination of phosphine in wheat by headspace gas-liquid chromatography. Journal of agricultural and food chemistry, 1978. 26(4): p. 962–4.
11. Nowicki, T.W., Gas-liquid chromatography and flame photometric detection of phosphine in wheat. Journal - Association of Official Analytical Chemists, 1978. 61(4): p. 829–36.
12. Berck, B., W.E. Westlake, and F.A. Gunther, Microdetermination of phosphine by gas-liquid chromatography with microcoulometric, thermionic, and flame photometric detection. Journal of Agricultural and Food Chemistry, 1970. 18(1): p. 143–7.
13. Longobardi, F., et al., Rapid Method for Determination of Phosphine Residues in Wheat. Food Analytical Methods, 2008. 1(3): p. 220–225.
14. Norman, K.N. and K. Leonard, Gas chromatography-mass spectrometry determination of phosphine residues in stored products and processed foods. Journal of agricultural and food chemistry, 2000. 48(9): p. 4066–70.
15. Robison, W.H. and H.W. Hilton, Gas Chromatography of Phosphine Derived from Zinc Phosphide in sugarcane. Journal of agricultural and food chemistry, 1971. 19(5): p. 875–878.
16. Martens-Menzel, R. and C. Reichmuth, Determination of phosphine in fumigated vegetables and in the air by headspace technique and GC. Gesunde Pflanzen, 1997. 49(6): p. 183–188.
17. Corley, J., et al., Rapid Zinc Phosphide Trace Analysis in Agricultural Commodities by Phosphine Generation, Toluene Trapping, and Gas Chromatography. Journal of Agricultural and Food Chemistry, 1998. 46(3): p. 999–1004.
18. Allen, S.E., Y.-L. Ren, and J.M. Desmarchelier, Comparison of six methods for determining aged phosphine residues in wheat. Journal of AOAC International, 1998. 81(3): p. 633–637.
19. Ren, Y.L., B. Padovan, and J.M. Desmarchelier, Evaluation of headspace solid-phase microextraction for analysis of phosphine residues in wheat. Journal of AOAC International, 2012. 95(2): p. 549–553.
20. Desmarchelier, J.M. and Y.L. Ren, Analysis of fumigant residues - A critical review. Journal of AOAC International, 1999. 82(6): p. 1261–1280.
21. Amstutz, R., A. Knecht, and D. Andrey, Detection of phosphine residues in organic cereals. Mitteilungen aus Lebensmitteluntersuchung und Hygiene, 2003. 94(6): p. 603–608.
22. Amrein, T., et al., Determination of phosphine in plant materials: Method optimization and validation in inter-laboratory comparison tests. Journal of agricultural and food chemistry. in print.
23. Berck, B., Sorption of Phosphine by Cereal Products. Journal of agricultural and food chemistry, 1968. 16(3): p. 419–425.
End of Article.
Share it: ![]()
![]()
![]()
![]()
